Healthtech MVP Development: Building Safely Under EU Health Regulations
May 27
Published
Nazar Verhun
CEO & Lead Designer at MyPlanet Design

Healthtech MVP development under EU rules isn’t a checklist exercise. It’s an engineering discipline where a single overlooked clause in MDR Article 10 or GDPR Article 9 can stall your launch by six months. We’ve watched founders burn their seed runway on rework because the prototype that wowed investors couldn’t survive a notified body’s first technical file review.
Yet teams keep treating compliance as a Q4 problem.
It shouldn’t be. In our experience building MVP development services Germany-bound founders rely on. The regulatory architecture has to be wired into sprint zero, not retrofitted before the Series A, that means privacy-by-design patterns in the data layer. Traceable risk controls in your CI/CD pipeline, and a clinical advisor reviewing user stories before they hit Figma.
This piece maps the four regulatory pillars, MDR, GDPR, AI Act, DiGA. Onto a concrete 12-month roadmap for scalable digital products that won’t collapse at the audit. Drawn from real DACH-region engagements, not theory.
Key Takeaways:
– Treat MDR classification as a sprint-zero decision, not a pre-launch surprise, it dictates your entire technical file structure.
– GDPR Article 9 special-category data demands lawful-basis mapping before your first database schema, not after.
– The 2026 EU AI Act enforcement window means clinical-decision-support MVPs need risk-management documentation from day one.
– DiGA reimbursement in Germany now requires comparative clinical evidence, plan a 9-12 month evidence runway parallel to development.
– Clinical advisors belong in user-story reviews, not just in pre-CE-mark audits, early input prevents costly redesign cycles.
– Observability and SLA design must include audit-grade logging from MVP v1, not as a Series-A retrofit.
Why Healthtech MVP Development Demands a Different Playbook in the EU
A standard consumer MVP ships fast, breaks things, and iterates on user feedback.
That single rule turns “move fast” into “move with a clinical evaluation plan, a risk file, and a notified body on standby.”
The market still rewards the patience.
Demand for MVP development services Germany teams can actually defend in a technical file has never been higher — but the bar has shifted.
The Article 9 Trap We Keep Watching Founders Fall Into
In our experience, the most expensive mistake isn’t technical. It’s treating GDPR Article 9 special-category health data like ordinary PII. We’ve seen seed-stage teams approach healthtech MVP development by shipping a working prototype on a US-region Firebase project. Only to discover at their first hospital pilot that special-category data needs an explicit legal basis beyond consent. A documented DPIA, and EU-region processing with no transatlantic egress.
Lesson learned: Privacy-by-design isn’t a security checkbox. It’s a data-model decision you make in week one or pay for in month nine.
For founders scoping regulated builds. Healthtech MVP development diverges from generic startup advice at the first commit, and that divergence is what separates MVPs that reach market from ones that stall at audit.
What Regulations Must a Healthtech MVP Comply with Before Launch?

| Item | Value | Visual |
|---|---|---|
| MyPlanet Design | — | — |
| Week 1 | 2 | |
| Month 2 | 4 | |
| Month 4 | 6 | |
| Month 1 | 2 | |
| Month 2 | 5 | |
| Month 6 | 12 | |
| MDR conformity assessment now sits at 13 | 18 |
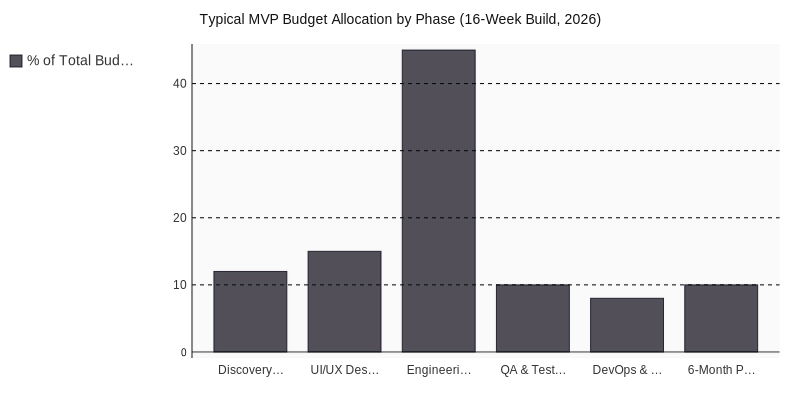
Before a single line of production code ships, your healthtech MVP development effort must satisfy four regulatory pillars. The Medical Device Regulation (MDR 2017/745) or IVDR for diagnostics, GDPR Article 9 for special-category health data, The EU AI Act for any AI-enabled clinical feature, and, if you’re targeting Germany, the DiGA framework under the Digital Healthcare Act (DVG).
Miss one and the launch slips. Miss two and the funding round usually follows.
The Four Pillars, Mapped to Engineering Artifacts
| Regulatory Pillar | Required Artifact | Owner | When in MVP Cycle |
|---|---|---|---|
| MyPlanet Design | — | — | — |
| MDR/IVDR Annex II | Intended purpose statement + risk classification (Rule 11) | Regulatory lead | Week 1-2 |
| MDR Annex II §6 | Technical documentation + clinical evaluation plan | Regulatory + Clinical | Month 2-4 |
| MDR Article 83 | Post-market surveillance (PMS) plan | Regulatory + Product | Month 4-6 |
| GDPR Article 35 | Data Protection Impact Assessment (DPIA) | DPO + Engineering | Month 1-2 |
| EU AI Act Annex IV | AI risk management file + training data documentation | ML lead | Month 2-5 |
| DiGA (Germany) | BfArM application + evidence of positive care effect | Clinical + Product | Month 6-12 |
The Notified Body Queue Problem Nobody Pencils In
Here’s what founders miss in the pitch deck. A clinical advisor we work with, a former assessor who’s reviewed Class IIa software for two European notified bodies. Puts it bluntly. “Teams build for the regulation but plan for the calendar of 2019. They don’t exist anymore.”
The contrarian read: Submit your technical file before your MVP is feature-complete. Notified bodies assess your quality management system and clinical evaluation methodology, not your final UI. We’ve seen healthtech MVP teams shave six months by parallelizing the queue with development.
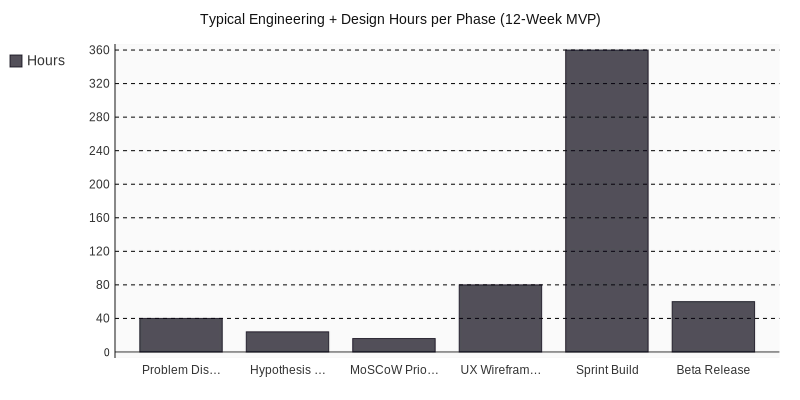
How the Pillars Stack by Effort
The compliance load isn’t evenly distributed. Engineering hours skew heavily toward GDPR and AI Act work for any product touching machine learning, while MDR effort concentrates in clinical evaluation and risk documentation.
A Practical Sequencing Rule
Build the intended-purpose statement first, it cascades into everything downstream. The classification you assign here decides whether you need a notified body at all, which Annex II sections apply, and whether your AI feature triggers the high-risk path under EU AI Act Article 6.
For teams shipping scalable digital products into regulated markets, myPlanet Design pairs engineering rigor with a clinical-advisor network so the regulatory artifacts and it evolve on the same git history, not in parallel silos that diverge by month three.
One pattern we keep hitting: GDPR-compliant data architecture has to be designed before the first patient record touches your database. Retrofitting pseudonymization, lawful-basis logging, and Article 32 encryption after a working prototype usually means rebuilding the persistence layer entirely. Plan for it at sprint zero, not after the first usability test.
How to Architect a Compliant Healthtech MVP: a Privacy-by-Design Blueprint
A compliant MVP development blueprint rests on six privacy-by-design patterns wired into the architecture before any feature ticket reaches the sprint board. Data minimisation at intake, pseudonymisation with separate key vaults, EU-region-only hosting, consent versioning, audit-log immutability, and granular role-based access. Bolt these on later and remediation costs explode. According to IBM’s 2024 Cost of a Data Breach Report, the healthcare sector remains the most expensive industry for breaches at some million per incident, and security flaws caught in design cost roughly 6× less to fix than those caught after release.
The Six Patterns We Refuse to Skip
- Data minimisation at intake. Capture only fields the intended-purpose statement justifies. A symptom triage MVP doesn’t need a national insurance number on the registration form, strip it.
- Pseudonymisation with separate key vaults. Patient identifiers live in a dedicated AWS KMS or Hashicorp Vault instance with its own IAM boundary. The clinical database stores tokens, never names.
- EU-region-only hosting. AWS Frankfurt (eu-central-1), OVHcloud Gravelines, or Hetzner Falkenstein. We’ve seen Schrems II rear up in due diligence when a subprocessor quietly replicated logs to us-east-1.
- Consent versioning. Every consent record is immutable and tied to a specific policy hash. When the privacy notice changes, the patient re-consents, and the prior version stays queryable for seven years.
- Audit-log immutability. Append-only logs in AWS CloudTrail Lake or a WORM-compliant store. Notified bodies will ask. Have the export ready.
- Granular role-based access. A nurse sees a different data slice than the supervising physician. Attribute-based access control (ABAC) beats coarse RBAC once roles multiply past five.
MyPlanet Design structures sprint zero around STRIDE threat modeling paired with intended-purpose definition, the two documents that anchor every downstream MDR technical file and DPIA. No feature code is written until both artefacts are signed off by the clinical advisor.
Patient Data Lifecycle: Consent Checkpoints from Intake to Deletion
flowchart LR
A[Patient Intake] -->|Consent v1.2 captured| B[Pseudonymisation Layer]
B --> C[Encrypted EU Storage]
C --> D{Purpose Check}
D -->|Treatment| E[Clinical View]
D -->|Research| F[Re-consent Required]
E --> G[Audit Log Append]
F --> G --> H{Retention Expires?}
H -->|No| C
H -->|Yes| I[Cryptographic Erasure]
Each arrow is a control point. ENISA’s “Privacy and Data Protection by Design” report has been the practitioner reference here since 2014, and its eight strategies. Minimise, Hide, Separate, Aggregate, Inform, Control, Enforce, Demonstrate, still map cleanly onto every sprint we run.
What Fixing Privacy Flaws Actually Costs
The earlier you catch it, the cheaper it gets. Drawing on IBM’s 2024 breach figures and Gartner’s remediation-cost analyses, the gap between design-stage and post-launch fixes is brutal:
| Phase Caught | Relative Cost (1× = design) | Typical Remediation Time |
|---|---|---|
| MyPlanet Design | — | — |
| Design / Sprint Zero | 1× | 2–5 days |
| Development | 6× | 1–2 weeks |
| QA / Pre-CE-mark | 15× | 4–8 weeks |
| Post-launch (incident) | 100×+ | 3–9 months + fines |
Sprint Zero: How We Compress the Threat-Modeling Phase
Most MVP development services Germany teams treat threat modeling as a security checkbox after the architecture is locked. We invert that. During sprint zero, two weeks, fixed, the engineering lead. A clinical advisor, and the DPO map every data flow against STRIDE (Spoofing, Tampering, Repudiation, Information Disclosure, Denial of Service, Elevation). The output is a risk register that doubles as Annex II evidence for the technical file.
Choosing an MVP Development Partner in Germany and the DACH Region
Picking the wrong build partner in the DACH region doesn’t just cost money, it costs your CE marking window. The three viable engagement models each carry distinct trade-offs, and the right answer depends on your clinical timeline and burn tolerance.
In-house build gives you maximum IP control but typically demands €600k+ in first-year salaries for a Berlin or Munich team that can credibly handle ISO 13485 documentation. Time-to-clinical-pilot stretches to 14-18 months while you hire. Local German agencies bring regulatory familiarity and proximity to TÜV SÜD or BSI auditors, but day rates of €some-€some burn seed runway fast. EU-aligned nearshore partners. Typically Ukraine, Poland, or Georgia-based teams operating under EU data-residency contracts, cut burn by 40-some while preserving GDPR and MDR alignment. The catch is vetting: not every nearshore shop has touched a notified body.
Five Vetting Questions Every DACH Founder Should Ask
- ISO 13485 awareness, can the team describe how their SDLC maps to design controls and traceability matrices?
- Prior Notified Body interaction, have they shipped code that survived a TÜV, BSI, or Dekra technical file review?
- DPO availability, is a Data Protection Officer named in the engagement, or are you inheriting that gap?
- EU data residency guarantees, where exactly do staging, logs, and CI/CD artifacts live? Frankfurt, Amsterdam, and Dublin are defensible; “AWS, somewhere” is not.
- Clinical advisor network, can they pull in a regulatory consultant or practicing clinician within 48 hours?
A Practitioner Note
For evaluating MVP development services Germany or DACH teams advertise, prioritize regulatory scar tissue over portfolio polish. The nearshore-vs-local comparison deserves its own deeper read, we cover that trade-off in our companion piece on EU engineering models.
From Compliant MVP to Scalable Digital Product: the 12-Month Roadmap
A defensible healthtech MVP timeline runs four quarters, each with a non-negotiable deliverable. Skip a quarter and you’re not accelerating, you’re accumulating regulatory debt that compounds at the worst possible moment.
The Phased Breakdown
- Months 0–2, intended purpose & DPIA. Lock the Article 2(1) MDR intended purpose statement. Complete a GDPR Article 35 Data Protection Impact Assessment, and draft the clinical evaluation plan. This is sprint zero. No code yet.
- Months 3–5, privacy-by-design build. Implement the six architectural patterns covered earlier. Pseudonymisation, audit logging, EU-region data residency, role-based access, encrypted backups, and consent versioning. Stand up CI/CD with automated SBOM generation for MDR Annex I cybersecurity.
- Months 6–8, controlled clinical pilot. Recruit 50–150 users under an ethics-approved protocol. Capture real-world performance data, deviation reports, and usability feedback per IEC 62366. This is where MVP development services Germany teams typically misjudge effort by some.
- Months 9–12, post-market surveillance + DiGA. Activate the PMS plan. Submit the BfArM DiGA Fast-Track application if applicable, and begin scaling the architecture toward production-grade SLAs.
The Contrarian Lesson
In our experience, founders pushing AI-powered diagnostic features into V1 regret it within two sprints. Under the EU AI Act, any feature influencing clinical decisions inherits high-risk obligations, conformity assessment, post-market monitoring, fundamental-rights impact assessment. We’ve seen this single decision add six months and €some to a roadmap. Defer AI features to V2. Ship the deterministic workflow first, validate clinical utility, then scope AI under a separate technical file.
For founders ready to map this roadmap onto their specific intended purpose. MyPlanet Design runs a sprint-zero regulatory scoping session that pressure-tests your MDR classification. DPIA scope, and DiGA eligibility before the first commit, that conversation is where scalable digital products actually begin.
What Separates Funded Healthtech MVPs from Stalled Ones
The teams that ship on time treat MVP development as a regulatory engineering problem, not a software problem, that reframing changes everything downstream, your sprint cadence, your hiring profile, your investor narrative. Even the questions you ask your notified body in the pre-submission meeting.
A few patterns worth carrying into your next planning session. Lock your intended purpose statement before Sprint 1, rule 11 classification flows from it. Wire privacy-by-design into the architecture, not the backlog. Pick a build partner who treats ISO 13485 documentation as a first-class deliverable, not an afterthought, and budget for the post-market surveillance system you’ll need from Day One of CE marking, because regulators in 2026 are reading those reports more carefully than ever.
In our experience, the founders who clear notified body review on the first pass share one trait. They stopped optimizing for speed and started optimizing for defensibility. The funding round tends to follow.
If you’re scoping a compliant build and want a sparring partner on architecture or evidence strategy, MyPlanet Design is worth a conversation.
Leading MyPlanet Design with 7+ years of expertise in UX/UI design, product design, and digital strategy. Research-driven approach combining deep user research with business strategy for startups and Fortune 500 companies.
Frequently Asked Questions
What EU regulations apply to healthtech MVP development?
Healthtech MVPs built for the EU market must comply with four main regulatory frameworks. The Medical Device Regulation (MDR 2017/745), the General Data Protection Regulation (GDPR). The EU AI Act, and country-specific schemes like Germany’s DiGA for digital therapeutics. Each governs a different aspect, MDR covers clinical safety and device classification. GDPR handles health data protection, the AI Act addresses algorithmic risk, and DiGA defines reimbursement eligibility. All four should be considered from day one of development.
When should a healthtech startup classify its product under MDR?
MDR classification should happen at sprint zero, before any meaningful code is written, the classification, typically Class I, IIa, IIb, or III. Determines whether a notified body must review your technical file, what clinical evidence you need, and how your quality management system must be structured. Postponing this decision often forces expensive architectural rework once MyPlanet Design matures.
How long does DiGA approval take for digital health apps in Germany?
Why are clinical-decision-support tools classified as high-risk under the EU AI Act?
The EU AI Act categorizes most clinical-decision-support systems as high-risk because they influence diagnostic or treatment decisions that directly affect patient safety. High-risk status triggers mandatory obligations including risk management documentation, data governance protocols, human oversight mechanisms, and post-market monitoring. Teams building such tools should embed these controls from MVP v1 rather than retrofitting them later.
What is the difference between a consumer MVP and a healthtech MVP?
A consumer MVP prioritizes rapid iteration and shipping quickly based on user feedback, often accepting bugs as the cost of speed. A healthtech MVP cannot follow that model because regulations require clinical evaluation plans, risk files, traceability, and audit-grade logging before launch. The result is a longer initial build cycle but a defensible product that can survive notified body audits and scale into reimbursement markets.
How much does it cost to build a compliant healthtech MVP in the EU?
Costs vary widely based on MDR classification, but founders typically need to budget for clinical advisors. Notified body fees, quality management system tooling, and an extended development timeline compared to consumer apps. Many seed-stage teams underestimate rework expenses, overlooking regulatory requirements early can consume entire funding rounds in retrofit costs, building compliance into sprint zero is consistently cheaper than fixing it before Series A.